Scientific advancements have often been held back by the need for high volumes of data, which can be costly, time-consuming, and sometimes difficult to collect. But there may be a solution to this problem when investigating how our bodies fight illness: a new machine learning method called "MotifBoost." This approach can help interpret data from T-cell receptors (TCRs) in identifying past infections to specific pathogens. By focusing on a collection of short sequences of amino acids in the TCRs, a research team achieved more accurate results with smaller datasets. This work may shed light on the way the human immune system recognizes germs, which may lead to improved health outcomes.
The recent pandemic has highlighted the vital importance of the human body's ability to fight back against novel threats. The adaptive immune system uses specialized cells, including T-cells, which prepare an array of diverse receptors that can recognize antigens specific to invading germs even before they arrive for the first time. Therefore, the diversity of the receptors is an important topic of investigation. However, the correspondence between receptors and the antigens they recognize is often difficult to determine experimentally, and current computational methods often fail if not provided with enough data.
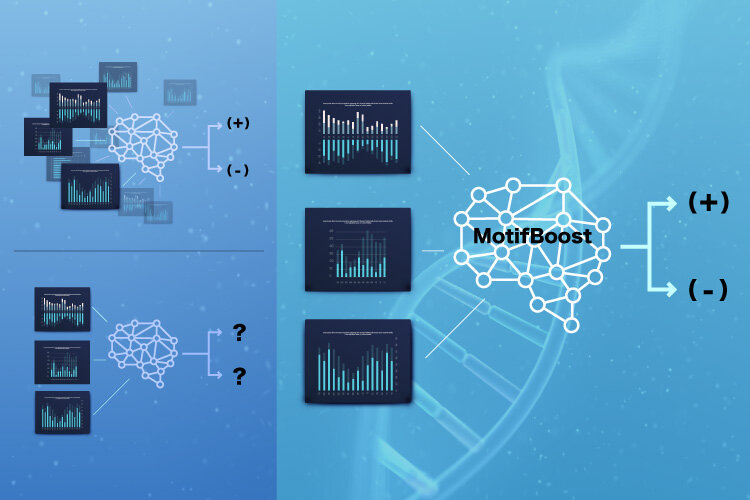
Now, scientists from the Institute of Industrial Science at The University of Tokyo have developed a new machine learning method that can predict the infection of a donor based on limited data of TCRs. "MotifBoost" focuses on very short segments, called k-mers, in each receptor. Although the protein motifs considered by scientists are usually much longer, the team found that extracting the frequency of each combination of three consecutive amino acids was highly effective. "Our machine learning methods trained on small-scale datasets can supplement conventional classification methods which only work on very large datasets," first author Yotaro Katayama says. MotifBoost was inspired by the fact that different people usually produce similar TCRs when exposed to the same pathogen.
First, the researchers employed an unsupervised learning approach, in which donors were automatically sorted based on patterns found in the data, and showed that donors formed distinct clusters using the k-mer distribution based on having previous infection by cytomegalovirus (CMV) or not. Because unsupervised learning algorithms do not have information about which donors had been infected with CMV, this result indicated that the k-mer information is effective in capturing characteristics of a patient's immune status. Then, the scientists used the k-mer distribution data for a supervised learning task, in which the algorithm was given the TCR data of each donor, along with labels for which donors were infected with a specific disease. The algorithm was then trained to predict the label for unseen samples, and the prediction performance was tested for CMV and HIV.
"We found that existing machine learning methods can suffer from learning instability and reduced accuracy when the number of samples drops below a certain critical size. In contrast, MotifBoost performed just as well on the large dataset, and still provided a good result on the small dataset," says senior author Tetsuya J. Kobayashi. This research may lead to new tests for viral exposure and immune status based on T-cell composition.
###
This research was published in Frontiers in Immunology as "Comparative study of repertoire classification methods reveals data efficiency of k-mer feature extraction" (DOI: 10.3389/fimmu.2022.797640).
Research Contact
Tetsuya J. Kobayashi, Associate Professor
Institute of Industrial Science, The University of Tokyo
Tel: +81-3-5452-6798
E-mail: tetsuya(Please add "@sat.t.u-tokyo.ac.jp" to the end)
URL:https://research.crmind.net/