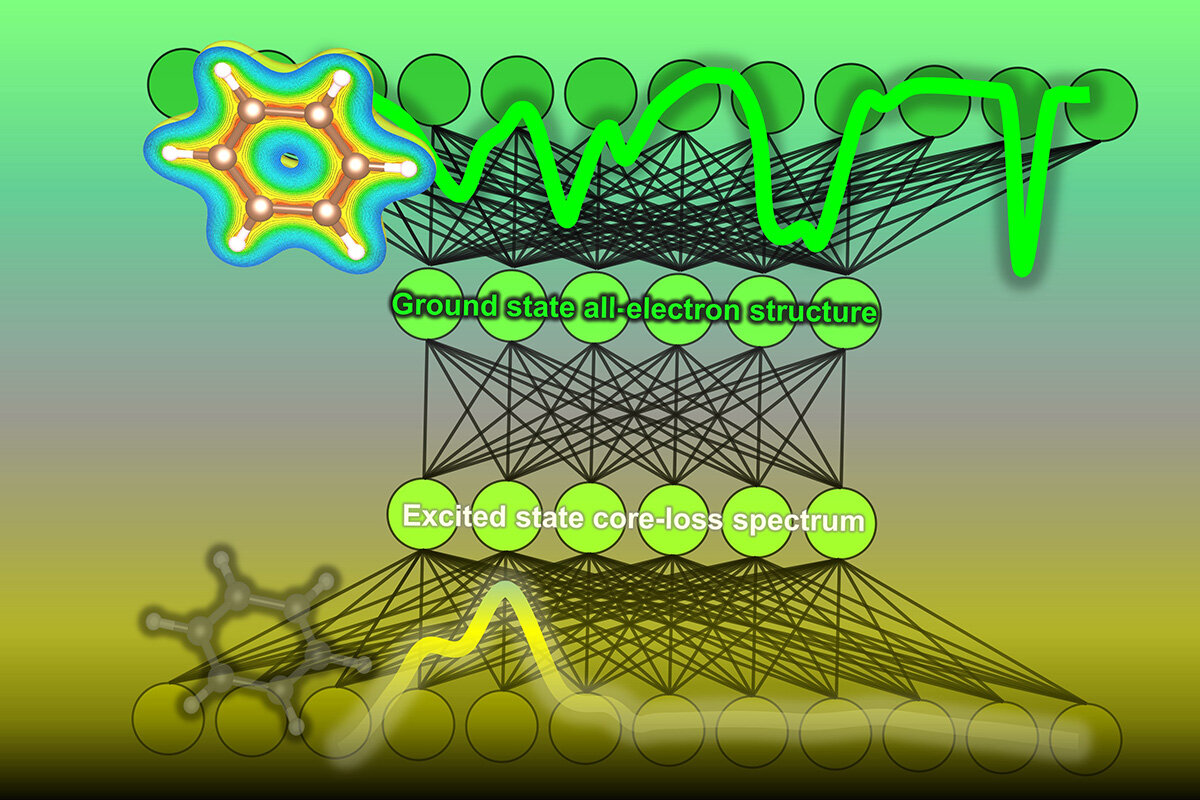
Organic chemistry, the study of carbon-based molecules, underlies not only the science of living organisms, but is critical for many current and future technologies, such as organic light-emitting diode (OLED) displays. Understanding the electronic structure of a material's molecules is key to predicting the material's chemical properties.
In a study recently published by researchers at the Institute of Industrial Science, The University of Tokyo, a machine-learning algorithm was developed to predict the density of states within an organic molecule, i.e., the number of energy levels that electrons can occupy in the ground state within a material's molecules. These predictions, based on spectral data, can be of great help to organic chemists and materials scientists when analyzing carbon-based molecules.
The experimental techniques often used to find the density of states can be difficult to interpret. This is particularly true for the method known as core-loss spectroscopy, which combines energy loss near-edge spectroscopy (ELNES) and X-ray absorption near-edge structure (XANES). These methods irradiate a beam of electrons or X-rays at a sample of material; the resulting scatter of electrons and measurements of energy emitted by the material's molecules allow the density of states the molecule of interest to be measured. However, information the spectrum has is only at the electron absent (unoccupied) states of the excited molecules.
To address this issue, the team at the Institute of Industrial Science, The University of Tokyo, trained a neural network machine-learning model to analyze the core-loss spectroscopy data and predict the density of electronic states. First, a database was constructed by calculating the densities of states and corresponding core-loss spectra for over 22,000 molecules. They also added some simulated noise. Then, the algorithm was trained on core-loss spectra and optimized to predict the correct density of states of both occupied and unoccupied states at the ground state.
"We attempted to extrapolate predictions for larger molecules using a model trained by smaller molecules. We discovered that the accuracy can be improved by excluding tiny molecules," explains lead author Po-Yen Chen.
The team also found that by using smoothing preprocessing and adding specific noise to the data, the predictions of density of state can be improved, which can accelerate adoption of the prediction model for use on real data.
"Our work can help researchers understand the material properties of molecules and accelerate the design of functional molecules," senior author Teruyasu Mizoguchi says. This can include pharmaceuticals and other exciting compounds.
The work, "Prediction of the Ground-State Electronic Structure from Core-Loss Spectra of Organic Molecules by Machine Learning," is published in The Journal of Physical Chemistry Letters at DOI: 10.1021/acs.jpclett.3c00142.
Research Contact
Teruyasu Mizoguchi, Professor
Institute of Industrial Science, the University of Tokyo
E-mail:teru (Please add "@iis.u-tokyo.ac.jp" to the end)